- Home
- Introduction
- Downloads
- Example
-
User Guide
Index
 System requirement Installation Memory configuration, reducing memory usage Updating annotation databases Main user interface Data inputs Creating a project Annotating a project Using user annotation track [GFF3/BED] Analyzing a project Selecting genes or regions Exome or targeted capture sequencing A command line tool Version history Updating to the latest version FAQ Requests & discussions License
System requirement Installation Memory configuration, reducing memory usage Updating annotation databases Main user interface Data inputs Creating a project Annotating a project Using user annotation track [GFF3/BED] Analyzing a project Selecting genes or regions Exome or targeted capture sequencing A command line tool Version history Updating to the latest version FAQ Requests & discussions License
- Screenshot
- Java Dev
- Plug-ins
- Visitors
Using this software
0. For impatient users1. Data inputs
2. Create a project
3. Annotate a project
4. Filter for quality scores
5. Main user interface
6. SVA genome browser
7. SVA tables
8. Selecting genes or regions
9. Analysis
10. Exome or targeted capture sequencing
A command line tool
FAQ
Requests and discussions

Memory configuration & Options to reduce memory usage
The minimum RAM requirement for running SVA is 32GB. However, we recommend 48GB or more RAM for more productive results.
Here are several notes for configuring RAM allocation for SVA:
(1) Please make sure your LINUX and JAVA are both 64-bit.
(2) After SVA is successfully launched, there is a menu item at "Options -> Memory Allocation". This menu brings up a dialog from where you can specify the amount of RAM allocated to SVA. Please do not allocate more than the amount the system can allow, otherwise the SVA won't start normally.
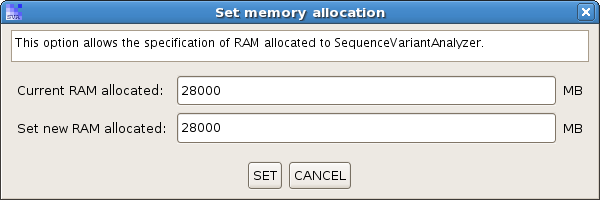
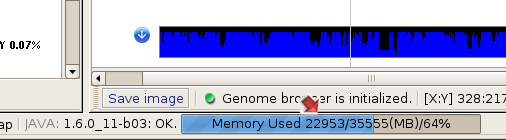
(3) SVA monitors its memory usage through a progress bar in the lower tool bar.
(4) There is a text file at: '[YOUR SVA DIR]/configuration/memory.txt' . Directly modifying the number in this text file (2nd line, units in MB) is equivalent to item (2) above.
Options to reduce memory usage
There are several options that users may consider to reduce the memory usage:
(1) Use less annotation threads
Users may click on menu item "Options -> Threads for annotation" and choose less computational threads to reduce the memory usage. This option will lead to slightly slower annotation speed but with less memory consumption.
(2) Split whole genome projects into chromosome-wise projects
(3) Use [MINIMUMMEMORY] option in the SVA .gsap script file.
Users may include a statement
[MINIMUMMEMORY] =ON
in their SVA .gsap project script to significantly reduce the memory usage. This option will
(3.1) restrict the core annotation to gene annotation and user-track annotation only. The reference variation database annotation will be skipped.
(3.2) use the minimum annotation thread.
| Visits: |
© 2011
© 2011