- Home
- Introduction
- Downloads
- Example
-
User Guide
Index
 System requirement Installation Memory configuration, reducing memory usage Updating annotation databases Main user interface Data inputs Creating a project Annotating a project Using user annotation track [GFF3/BED] Analyzing a project Selecting genes or regions Exome or targeted capture sequencing A command line tool Version history Updating to the latest version FAQ Requests & discussions License
System requirement Installation Memory configuration, reducing memory usage Updating annotation databases Main user interface Data inputs Creating a project Annotating a project Using user annotation track [GFF3/BED] Analyzing a project Selecting genes or regions Exome or targeted capture sequencing A command line tool Version history Updating to the latest version FAQ Requests & discussions License
- Screenshot
- Java Dev
- Plug-ins
- Visitors
25 Jan 2012:: We now have a new download site: www.OmicsExpress.com
25 May 2011:: SVA is published in Bioinformatics.
21 Mar 2011:: SVA V1.10 is released: [1]Supports GRCh build 37/hg19; [2] Supports user annotation track in GFF3 or BED formats.
9 Sep 2010:: The characterization of twenty sequenced human genomes. [Article]
12 Jul 2010:: LabCorp Launches Interleukin 28B Polymorphism (IL28B) Genotype Test to Support Individualized Treatment Decisions for Patients with Hepatitis C Viral Infection.
17 Jun 2010:: Causal variants for metachondromatosis are identified.
[Article] [SVA screenshot]
[GenomeWeb: The Daily Scan]
[Article] [SVA screenshot]
{kind=link}
[GenomeWeb: The Daily Scan]
18 Mar 2010:: SVA 1.02[beta] is released.
11 Mar 2010:: SVA 1.01[beta] is released with a command line tool.
8 Mar 2010:: A lite evaluation edition is released for Windows. Play with it on your laptop!
23 Jan 2010:: SVA 1.00[beta] is released.

How does SVA work?
SVA uses a number of biological databases to perform the functional annotation, and then, implements several internal and external programs to perform the statistical and bioinformatical analyses for identifying potential causal variants and genes responsible for the biological traits or medical outcomes of interest.
The chart below may give you a brief idea on what resources SVA uses and what information SVA implements to compile the annotation and analyses. There is also a list of these resources at the end of page.
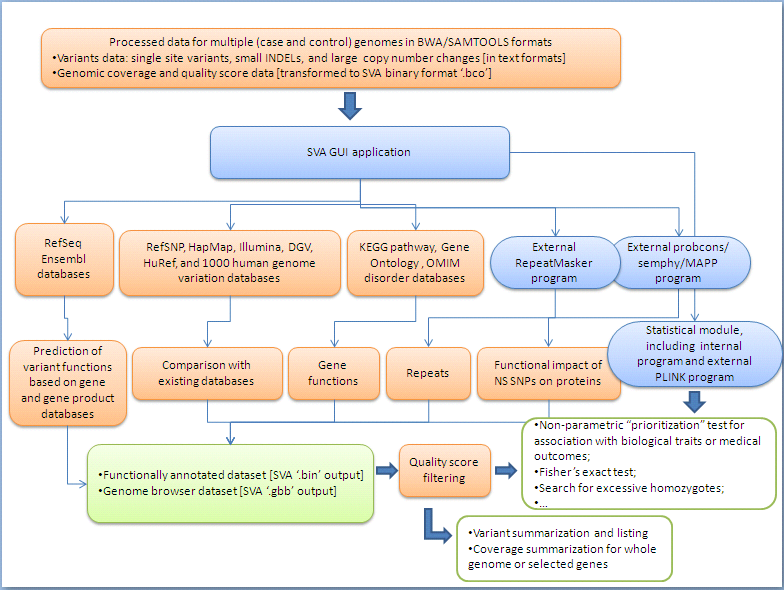
Resources used by SVA:
[Note]: only (small) part of each the following resources were implemented in SVA for performing the annotation and analyses.
- Ensembl databases: Ensembl is a joint project between EMBL - EBI and the Wellcome Trust Sanger Institute to develop a software system which produces and maintains automatic annotation on selected eukaryotic genomes. [Reference: Flicek P, Aken BL, Ballester B, et al. Ensembl's 10th year. Nucleic Acids Res 2009. ]
- NCBI RefSeq: The Reference Sequence (RefSeq) collection aims to provide a comprehensive, integrated, non-redundant, well-annotated set of sequences, including genomic DNA, transcripts, and proteins. RefSeq is a foundation for medical, functional, and diversity studies; they provide a stable reference for genome annotation, gene identification and characterization, mutation and polymorphism analysis, expression studies, and comparative analyses.
- NCBI dbSNP : The dbSNP database.
- HapMap: The International HapMap Project is a multi-country effort to identify and catalog genetic similarities and differences in human beings. Using the information in the HapMap, researchers will be able to find genes that affect health, disease, and individual responses to medications and environmental factors. The goal of the International HapMap Project is to compare the genetic sequences of different individuals to identify chromosomal regions where genetic variants are shared. [Reference: The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851-861. 2007. ]
- Illumina Infinium HD Human1M BeadChip: a commercial GWAS (genome-wide association studies) genotyping chip manufactured by Illumina Inc.
- DGV: A curated catalogue of structural variation in the human genome. [Reference: Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet 2004; 36 (9) : 949-51. ]
- HuRef: The diploid genome sequence of J. Craig Venter. [Reference: Levy S, Sutton G, Ng PC, et al. The diploid genome sequence of an individual human. PLoS Biol 2007; 5 (10) : e254. ]
- The 1000 human genome project: The 1000 Genomes Project , launched in January 2008, is an international research effort to establish by far the most detailed catalogue of human genetic variation . [Note: only portion of the preliminary variation data were implemented by SVA.]
- KEGG pathway: KEGG pathway is a collection of manually drawn pathway maps representing knowledge on the molecular interaction and reaction networks for certain biological processes. [Reference: Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 2009. ]
- Gene ontology: The Gene Ontology project is a major bioinformatics initiative with the aim of standardizing the representation of gene and gene product attributes across species and databases.
- OMIM: Online Mendelian Inheritance in Man.
- RepeatMasker: RepeatMasker is a program that screens DNA sequences for interspersed repeats and low complexity DNA sequences.
- PROBCONS: Probabilistic Consistency-based Multiple Alignment of Amino Acid Sequences.
- SEMPHY: Structural EM Phylogenetic Reconstruction.
- MAPP: Multivariate Analysis of Protein Polymorphism.
- : Whole genome association analysis toolset.
Resources upstream to SVA:
- BWA: Burrows-Wheeler Aligner (BWA) is an efficient program that aligns relatively short nucleotide sequences against a long reference sequence such as the human genome.
- SAMtools: SAM Tools provide various utilities for manipulating alignments in the SAM format, including sorting, merging, indexing and generating alignments in a per-position format.
| Visits: |
© 2011
© 2011